人类Usher综合征的治疗方法:小分子忽略停止信号
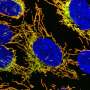
 capacity for bundling actin filaments (red). Photo: Tobias Goldmann")
亚瑟综合征是人类中最常见的先天性聋人失明的形式,并影响6,000人的人口。它是一种隐性遗传疾病,临床和遗传异质性。在最严重的情况下,患者出生聋,开始患有青春期的视网膜的退化,最终导致完全失明。这些患者在日常生活中经历了重大问题。在听力损失可以用助听器和耳蜗植入物补偿,但迄今为止没有证明可以为相关视力丧失制定治疗。Johannes Gutenberg大学Mainz(JGU)的研究人员现在已经为这种疾病制定了一种新的治疗方法。
美因茨大学动物研究所的乌韦·沃尔夫姆教授领导的研究小组在之前的研究中已经深入了解了这一基本原理分子过程以及导致这种衰弱综合症的机制。利用这项成功的基础研究结果,美因茨的Usher治疗小组由Kerstin Nagel-Wolfrum博士领导,现在已经评估了潜在的眼部治疗方案。他们的注意力集中在一个特定的德国家庭中发现的一种突变上,该家庭已知会导致最严重的鄂榭综合症。这种突变是USH1C基因中所谓的无义突变,导致在DNA碱基中产生停止信号,导致过早终止蛋白质合成。
Mainz研究团队现已发布其最新的药物发生策略,用于治疗亚瑟综合征患者无意义突变在五月版的杂志上人类基因治疗。研究人员发现,一种名为PTC124 (Ataluren)的小分子可以使突变的USH1C基因中的停止信号被忽略,从而导致蛋白质的持续合成,并在细胞和器官培养中形成功能性遗传产物。除了能够引起停止信号的读取,活性药物PTC124也被证明与小鼠和人类视网膜培养物高度兼容。此外,该团队首次成功地在体内演示了眼睛突变密码子的通读。
“PTC124已经在临床试验中进行了测试,以其在治疗涉及非礼突变的其他疾病中的疗效,例如囊性纤维化和杜氏肌营养不良症。我们希望这种治疗方法很快就可以在亚瑟综合征患者中使用,”博士解释说Kerstin Nagel-Wolfrum。
托拜厄斯·戈德曼(Tobias Goldmann)目前正在对他的博士论文进行最后的润色,他正在比较通读率的效率和其他诱导无义突变通读的分子的生物相容性。重点是修饰氨基糖苷类,即商业上可获得和临床测试的抗生素衍生物。这些基因是由以色列的合作伙伴,海法技术中心的Timor Bassov教授设计和合成的,并且已经被美因茨的研究人员成功地用于解读Usher基因的无义突变。除了对这些新物质在眼部的应用进行进一步的临床前研究外,美因茨的Usher实验室正计划使用这种新的方法来治疗这种特殊形式的迎来综合征在医院病人尽快。
Goldmann T.,覆盖N.,Wolfrum U.,Nagel-Wolfrum K.(2011):PTC124介导的非义突变突变导致亚瑟综合征1C型的非义突变升高。人类基因治疗22:537 - 547。DOI: 10.1089 / hum.2010.067
Goldmann T., Rebibo-Sabbah A., Overlack N., Nudelman I., Belakhov V., Baasov T., Ben-Yosef T., Wolfrum U., Nagel-Wolfrum K.(2010):在视网膜中设计氨基糖苷NB30的USH1C无义突变的有益解读。调查眼科与视觉科学51:6671 - 80。DOI:10.1167 / iovs.10-5741












用户评论