生物学家实现了负责迎膜综合征的停止突变的修复和接通
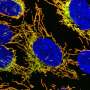
. In the presence of NB54, there is read-through of the USH1C mutation. The treated cells produce the healthy harmonin protein (green). Credit: Kerstin Nagel-Wolfrum")
经过多年的基础研究,美因茨约翰内斯古登堡大学(JGU)的科学家们越来越能够理解人类Usher综合征的潜在机制,并越来越接近于找到一种成功的治疗方法。乌威·沃尔夫鲁姆(Uwe Wolfrum)教授的亚瑟小子研究小组的科学家们正在评估两种不同的策略。这些方法涉及突变基因的修复或基因缺陷的失活。根据迄今获得的结果,这两种选择似乎都很有希望。Usher综合征是一种先天性疾病,会导致听力和视力的丧失。
Usher综合征是人类最常见的先天性聋哑人,发病率为每6000人中就有1人。患有这种疾病的人在日常生活中严重残疾,因为他们失去了使用两个最重要的感觉器官,即他们的耳朵和眼睛。在最严重的情况下,患者天生耳聋,并开始遭受视力损害的形式视网膜变性在青春期会导致完全失明。虽然有可能弥补听力的损失助听器和耳蜗植入设备在美国,以前没有治疗该疾病的眼部成分的方法。美因茨大学的科学家们目前正在进行临床前转化研究,试图找到这个问题的答案。
Kerstin Nagel-Wolfr博士团队的调查专注于USH1C基因的无意义突变,这些基因被确定为德国家庭中最严重的亚瑟患者综合征的原因。无意义突变是由DNA产生的止动信号,其导致蛋白质HarmunIn的合成的过早终止,其由USH1C编码。
该研究团队在六月型杂志调查视网膜学和视觉科学的举行的迎膜综合征中发表了最新调查结果。在她的博士研究期间,诺拉博士在使用所谓的锌 - 手指核酸酶技术的帮助下,诺拉博士通过分子剪刀的帮助来修复USH1C基因。使用锌手指核酸酶,科学家首先在疾病产生突变的部位发起双序列DNA切割。然后通过细胞自身的修复机制以同源重组的形式和引入非突变的USH1C DNA序列来修复该分子水平的外科切口。因此,突变的基因序列用非突变序列替换。在基因组和蛋白质水平的细胞培养模型中,在细胞培养模型中证明了锌指核酸酶技术关于遗传修复的效果。
该研究团队最近还在杂志上发表了其治疗乌谢尔综合征无义突变患者的药物遗传学方法的最新结果EMBO分子医学。在这种情况下,托拜厄斯·戈德曼博士和其他团队成员比较了各种可以诱导读取停止信号的分子,从而提供正常的蛋白质合成。此外,他们还评估了不同分子的视网膜生物相容性。研究集中在PTC124 (Ataluren®)和“设计”氨基糖苷类。这些氨基糖苷类物质来源于经过临床测试的抗生素,并由以色列海法技术中心的Timor Bassov教授对其进行了改良,以提高其识别突变并降低毒性的能力。美因茨的研究人员已经成功地使用第一代设计氨基糖苷类药物来解读USH1C基因的无义突变。
它们现在能够显示PTC124(Ataluren®)和第二代氨基糖苷(NB54),特别是在突变的USH1C基因中诱导止动信号的读取信号。这意味着蛋白质合成续,因此在细胞和器官培养物中合成活性基因产物。活性物质,PTC124和NB54,通常增强读取功效,并在与临床使用的抗生素相比,在小鼠和人视网膜培养物中表现出改善的耐受性。该团队还成功记录了Vivo鼠标模型的突变读取。
“我们基于基因的治疗策略,包括基因修复和通读疗法,代表了有价值和有前途的替代病毒基因添加,实际上可能是大型和亚型丰富的USH基因的唯一治疗选择。我们希望这些替代方案将对Usher综合征患者以及其他患有严重遗传性视网膜病变和其他遗传性疾病的患者的治疗做出重大贡献,”Kerstin Nagel-Wolfrum博士解释说。
除了继续其对活性物质使用的临床前研究,美因茨亚瑟研究团队计划使其新的迎来综合征尽快为患者提供治疗。
Tobias Goldmann等人,“NB30,NB54和PTC124的比较评估,用于治疗USH1C无意义突变的疗效”,EMBO分子医学, 2012年10月。doi: 10.1002 / emmm.201201438










用户评论