神经系统疾病的遗传原因确认
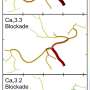
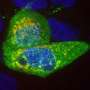
 changes the way the protein (green) opens and closes a pathway through neurons' cell membrane (blue). This change alters the way Calcium (Ca+) moves between the outside and inside of cells and is responsible for patients' disease symptoms. Credit: Hiroshima University")
研究人员利用两个三代患病的不同家庭的遗传信息,确定了一种导致退化和最终致命的运动障碍的新突变。通过诱导多能干细胞技术,研究人员还在实验室培育了一名患者的神经元,用于未来的实验。
Spinocerebellar Ataxia(SCA)是一种遗传疾病,导致浪费小脑,这部分脑的负责控制自愿肌肉运动,就像走路,说话,甚至是我们眼睛的方向一样。
目前,SCA没有治愈或治疗方法。的突变大约30%的病例仍未查明。
两个不同的SCA家庭在日本的两家不同的医院寻求治疗。在对有症状的个体进行初步测试后,医生没有发现任何已知的基因突变。广岛大学的研究人员随后收到了患者的基因样本,并开始了寻找新突变的过程。
经过SCA的四个家庭成员的遗传测序后,由Hideshi Kawakami教授,博士学位领导的研究小组从广岛大学流行病学系开始使用统计分析,将家庭的DNA与无关的人的DNA与没有SCA的无关人员。这种统计分析允许研究人员识别哪些遗传与SCA的家庭成员分享健康人没有。
导致这两个家族SCA的基因位于17号染色体上。这种名为CACNA1G的基因编码Cav3.1蛋白质。Cav3.1是神经细胞内部和身体其他部分之间的一种离子通道或通道。不同研究领域的科学家已经知道,当神经发出电脉冲时,Cav3.1可以控制进入神经的钙离子数量。Cav3.1之前从未与SCA关联过。
改变CACNA1G DNA序列中的一个字母,就可以改变2377个氨基酸链中的一个氨基酸,这些氨基酸是细胞连接起来构建Cav3.1蛋白质的。
研究人员进行了实验,以检查突变的Cav3.1通道在培养皿中生长的细胞中的行为方式。这种突变使得Cav3.1通道以较低的阈值打开,这意味着它们让钙进入细胞的方式不同于健康细胞。
“将来,修改该频道的药物可能会治愈患者,”Kawakami教授说。
一名患者的皮肤细胞被用于诱导多能干细胞在实验室里培育病人的神经元。这些新生神经元未表现出明显的躯体畸形,可能与SCA正常进展相符。根据SCA的突变类型,一些患者可能到中年才会出现症状。
“我们可能需要一些与年龄相关的因素来繁殖类似生活的细胞行为,”Kawakami教授说。
研究人员计划在未来的实验中使用这些神经元,在更逼真的条件下,对致病的Cav3.1进行更详细的研究。
进一步探索










用户评论