从人IPS细胞中复制多囊肾疾病的病理生理学

由日本熊本大学领导的联合研究,在体外成功地从人类iPS细胞中复制了常染色体显性多囊肾病(ADPKD)的发病机制。ADPKD是一种引起双侧肾脏多发囊肿的疾病,是最常见的遗传性肾脏疾病。虽然先前有关于肾小管囊肿的文献记载,但这是首次由集合管诱导的囊肿,这与疾病的发病机制更密切相关。研究人员预计,这将有助于更好地了解疾病状态,并开发新的治疗方法。
估计ADPKD每400-1000人中会影响1。虽然ADPKD患者的一半进展了肾功能衰竭到60岁时,这种疾病的潜在机制尚不清楚,治疗方法也尚未找到。ADPKD是由突变在PKD基因如果从父亲和母亲各自遗传的PKD基因的一个或两个副本发生突变(杂合或纯合突变),就会发展。PKD基因由PKD1和PKD2组成,约85%的ADPKD患者存在PKD1基因的杂合突变。剩下的15%有PKD2基因的杂合突变。PKD1基因杂合突变的患者预后更严重,大约一半的患者在60岁时由于肾脏的进展而发展为肾功能衰竭囊肿。ADPKD的机制主要研究小鼠。然而,具有PKD1基因中杂合突变的小鼠在成年人中发育了少数肾囊肿,并且不会再现人类症状。
肾脏的发育是通过两种不同的前体之间的相互作用;肾元祖和输尿管芽。肾元祖分化成肾小管,重新吸收尿液中的盐和水,输尿管芽分化成集尿管,收集尿液和重新吸收水。ADPKD囊肿可起源于肾小管和集合管,但集合管似乎是其主要来源。2014年,Nishinakamura等报道了一种从人类iPS中诱导肾小管的方法细胞通过肾元祖细胞。从那时起,几个研究小组已经成功地从PKD1基因纯合子突变的iPS细胞中培育出肾小管来源的囊肿,但尚未培育出集合管来源的囊肿。由于没有基因突变的正常iPS细胞也形成了肾小管囊肿,因此PKD1基因杂合突变的ADPKD患者的iPS细胞不可能复制疾病状态。然而,西中村教授的研究小组在2017年通过输尿管芽诱导了人类iPS细胞的收集管,并一直试图通过建立自己的方法来复制收集管衍生的囊肿。
-
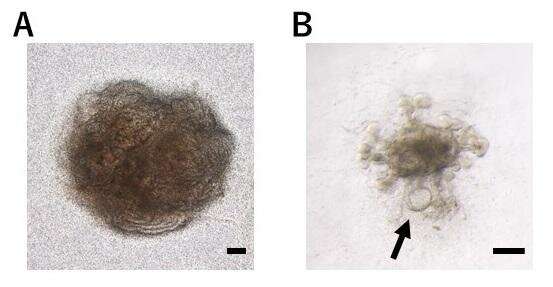
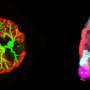
答:施用衍生自IPS细胞与PKD1纯合突变的肾小管施用的血管加压素不形成囊肿。(比例栏:200μm)学分:Ryuichi Nishinakamura教授 -
肾脏产生的尿液通过肾小管,在集尿管中收集,然后从体内排出。资料来源:Ryuichi Nishinakamura教授
为了再现ADPKD囊肿,研究人员使用基因编辑技术CRISPR-CAS9与PKD1基因创造纯合和杂合突变体IPS细胞,并将它们诱导成肾小管。在给予一种叫做苏克林的药物后,激发了加剧ADPKD囊肿的因素,如先前的报告中那样再现了管状囊肿。然而,来自尚未遗传突变的肾小管也形成的温和囊肿。
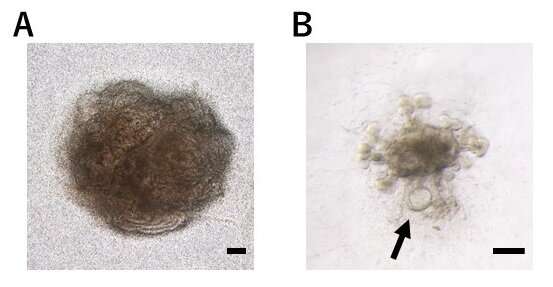
当将这些IPS细胞诱导收集管道并用Forskolin处理时,由具有PKD1纯合突变的收集管道形成的囊肿。另一方面,囊肿没有从没有基因突变的管道中形成的,表明它们对Forskolin的反应与肾小管的反应不同。当研究人员检查受体的表达时为抗毒激素加压素,其作用于收集管道,它们发现它仅在收集管道中表达。已知血管加压素可从收集管道加剧ADPKD囊肿,并且当施用于诱导的收集管道和肾小管时,形成的囊肿,尽管以低频,但仅在用PKD1纯合突变收集管道。此外,囊肿部分地形成在给予Forskolin后的PKD1杂合突变的管道中。当使用PKD1杂合突变的ADPKD患者的IPS细胞用于诱导收集管道,以相同方式形成囊肿。根据研究人员,这是来自患者衍生的IPS细胞的ADPKD疾病病理学的第一次成功再现。
“该研究表明,诱导IPS细胞收集管道的方法可以是一个新的疾病adpkd的型号,“基督教教授的研究领导者说。”通过分析这些收集管囊肿 - 这与实际临床状况类似 - 我们可能会发现机制并开发难以识别的新治疗方法。我们还期望从患者衍生的IPS细胞中复制囊肿将导致个体病例的研究和治疗。“
进一步探索











用户评论