研究人员利用病人的细胞测试罕见眼病的基因疗法

美国国家眼科研究所(NEI)的科学家已经开发出一种很有希望的基因治疗策略,用于治疗一种导致儿童严重视力丧失的罕见疾病。该疾病是一种莱伯氏先天性黑蒙,是由CRX基因常染色体显性突变引起的,用基因治疗具有挑战性。科学家们用实验室制造的由病人细胞制成的视网膜组织来测试他们的方法,这些组织被称为视网膜类器官。这种方法涉及在其自然控制机制下添加正常基因的副本,部分恢复了CRX的功能。研究报告发表在今天的干细胞的报道。NEI是美国国立卫生研究院的一部分。
NEI神经生物学、神经退行性和修复实验室主任、该报告的资深作者Anand Swaroop博士说:“我们的治疗方法,即添加更多正常基因的拷贝,有可能治疗由各种突变引起的常染色体显性LCA。”
美国食品和药物管理局(fda)于2017年批准Luxturna用于治疗RPE65基因突变的LCA患者。尽管Luxturna被誉为基因治疗的重大进展,但它对其他形式的LCA无效,包括那些由CRX常染色体显性突变引起的LCA。
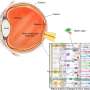
CRX基因编码一种蛋白质(也称为CRX),它与DNA结合,并指示视网膜的光感受器产生称为视蛋白的光敏色素。没有功能性CRX蛋白,光感受器就失去了探测光线的能力,最终死亡。
像常染色体显性LCA这样的疾病很难用基因疗法治疗,因为添加更多的正常基因并不总是能恢复功能。有常染色体显性突变的人仍然有一个正常的基因拷贝,但是突变的蛋白质会干扰正常的蛋白质。有时,简单地添加更多的正常蛋白质,而不是恢复正常功能,可能会以不可预测的方式加剧疾病。
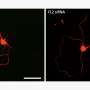
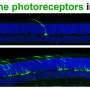
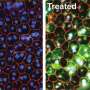
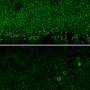
为了探索基因扩增——添加正常基因的复制品——如何影响常染色体显性LCA, Swaroop的团队从两名LCA患者及其未受影响的家庭成员身上开发了视网膜类器官。在卡米尔·克鲁切克博士(斯瓦鲁普实验室的博士后研究员)的带领下,他们分几个阶段构建了复杂的视网膜样组织,从皮肤细胞开始,利用每个志愿者的基因图谱诱导成熟的光感受器和其他视网膜细胞的产生。正如预期的那样,患者类器官产生的感光视蛋白远少于未受影响的家族成员所产生的类器官。
为了仔细控制受体光感受器会表达多少CRX基因,研究小组重新设计了CRX启动子,使其可以与CRX基因一起传递,作为基因治疗的一部分。启动子是一个邻近的DNA序列,它控制时间和方式基因表达。研究人员将该基因和他们的工程启动子包装在一种病毒内,这种病毒将它们运送到有机感光器中。
研究小组的基因扩增策略恢复了这两例患者的一些类器官的CRX蛋白功能,驱动两种类型的光感受器的视蛋白表达:视杆细胞和视锥细胞。
“事实上,这种策略对两种CRX突变都有效,这非常令人兴奋,”Swaroop说。“基因扩增可能是可行的治疗其他常染色体显性LCA突变。"
“这个概念基因治疗这项研究是为一种罕见LCA的潜在治疗方法迈出的第一步,”NEI临床主任和该研究的合著者Brian Brooks医学博士说。“这是基础和临床科学研究人员合作的一个很好的实验到临床科学的例子。”
进一步探索










用户评论