研究人员识别细胞缺陷家族和零星的常见形式的肌萎缩性侧索硬化症

肌萎缩性脊髓侧索硬化(ALS)是一种快速进步和致命退行性疾病影响大脑和脊髓神经细胞负责控制随意肌运动。“零星”或non-inherited ALS,约占90%的情况下,和10%的病例是由于已知的基因突变。通过研究实验室培养的神经元来自皮肤或血液细胞从10正常对照组,八ALS引起突变,和17 non-inherited ALS,研究人员发现一个可能的起点功能障碍导致的疾病。这项研究发表在科学转化医学是部分由国家神经疾病和中风研究所(研究所),美国国立卫生研究院的一部分。
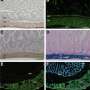
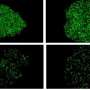
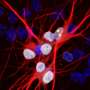
使用ALS patient-derived图书馆细胞Jeffrey Rothstein领导的研究小组,医学博士博士,约翰霍普金斯大学医学院的巴尔的摩,发达的诱导多能干细胞(iPSC)派生从病人的神经元培养细胞,发现了一个共同缺陷无论细胞来自人与继承或non-inherited肌萎缩性侧索硬化症。他们的报告,在ALS神经细胞,一种叫做CHMP7的蛋白质的积累在体外培养的神经细胞内细胞核以及ALS样本中控制运动的大脑区域。治疗,减少的数量在培养细胞CHMP7阻止ALS的一系列的异常特征。
“有相当大的兴趣识别治疗ALS的新目标,特别是对的零星的形式障碍,“说天使爱美丽Gubitz,博士项目负责人,研究所。这里显示的“基因打靶策略现在允许我们从生物发现直接治疗的发展。”
本研究建立在早期的论文Rothstein实验室,看着ALS的最常见的遗传原因,C9orf72基因的突变(也称为“C9突变”)。那里,他们表明C9突变产生缺陷的结构称为核孔隙负责移动蛋白质和其他分子的细胞核的细胞。具体地说,他们发现某些蛋白质缺席的毛孔,导致整个孔隙分裂domino-like效应。
“我们知道从我们以前的工作,C9突变产生核孔缺陷,但是我们不知道为什么,“Rothstein博士说。“在这里,我们要回答的问题发生了什么上游的孔隙缺陷通过研究神经元的细胞中提取出来的ALS患者。”
具体地说,研究人员观察了神经细胞从诱导多能干细胞(万能),这是一种干细胞,可以创建从一个人的皮肤或血液样品。这些细胞的行为很像其他干细胞,它们可以变成许多不同类型的细胞在实验室设置,包括神经细胞。通过与回答ALS,全国ALS生物数据和iPSC努力由Rothstein,研究人员能够访问则来自家族和零星的ALS患者。
“万能的优势之一是,您可以看看不同的时间非常相同的方式你会研究动物模型在不同的年龄,“Rothstein博士说。“我们知道核孔的时间点开始降低,我们可以研究神经元在早期的原因可能是。”
他们发现,核内的积累CHMP7发生至少一个星期前核孔的发展异常。通常,CHMP7迅速移除一旦进入细胞核,但在制备过程和零星的ALS iPSC-derived神经元,持续积累。如果一个反义寡核苷酸药物,阻止细胞制造特定的蛋白质,被用来减少在ALS CHMP7神经元的数量,毛孔不会退化。最后,如果一种变异的CHMP7无法从细胞核是添加到健康的神经元,孔隙退化就像在ALS神经元,这表明存在CHMP7核内神经元可能是一个关键事件发展的疾病。
一个异常常见的所有形式的ALS是另一种蛋白质的定位错觉,TDP-43。通常存在于细胞核,TDP-43泄漏到周围的细胞质在ALS聚合的聚合,导致的损失函数变化的各种类型的RNA,这对某些基因的翻译是至关重要的蛋白质。最终在iPSC-derived这也见过神经元制备过程和零星的ALS患者。CHMP7的反义寡核苷酸治疗后,TDP-43定位错觉,再也没有出现,RNA缺陷都得到纠正。
“这些发现让我们一起把这些异常序列,原子核中的CHMP7积累导致的地方核孔损伤,其次是TDP-43定位错觉,最终细胞死亡,“Rothstein博士说。“这不仅是局限于C9突变;它是零星的ALS的基本途径,可以用反义寡核苷酸CHMP7。”
Rothstein博士的实验室目前正在调查反义寡核苷酸药物是否可以发展成为一种治疗技术和零星的ALS患者。他们还继续研究的初始积累CHMP7确定什么导致了核的定位错觉。