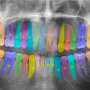
Double-stranded RNA induces bone loss during gum disease
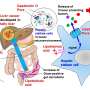
] analogue induces the PGE<sub>2</sub>-mediated expression of RANKL that stimulates osteoclast formation and directly prolongs the life span of mature osteoclasts, that was leading alveolar bone resorption in periodontal disease. Credit: Masaki Inada, Tokyo University of Agriculture and Technology")
Tokyo University of Agriculture and Technology researchers reported on a new discovery regarding the mechanisms for bone loss in gum disease (periodontitis). They found that double stranded RNA molecules can activate the immune system response that leads to deterioration of bone.
They published their paper in the March issue ofJournal of Biological Chemistry.
Serious gum infections damage soft mouth tissues such as gums and gradually erode the underlying (alveolar) bones that support our teeth. Both the bone pockets around the base of teeth and the ligaments anchoring teeth to the jawbone are susceptible to getting broken away bybacterial infection. This periodontal bone erosion, gone unchecked, may finally result in tooth loss.
It has long been recognized that concentrations of bacterial plaque nestled in the tooth pockets are the cause of periodontal disease. The main components of outer membranes of the bacteria that causegum diseaseare molecules called lipopolysaccharides. Lipopolysaccharides support the bacterial cell and protect against attack of immune cells, but have also been implicated in causing gum inflammation by switching on人数,like receptors(TLR4) on immune cells that then recognize the bacteria as pathogens.
However, until now it was unclear whether "other pathogens including double-stranded RNA (dsRNA) derived from bacteria or autologous cells contribute to the progression of periodontalbone loss," explains study author and professor Masaki Inada, D.D.Sc and Ph.D. in the Department of Biotechnology and Life Science. For example, immune cells such as neutrophils accumulated in inflammatory tissues could release dsRNA in the mouth. The recent study investigated dsRNA as a suspect in the progression of bone inflammation during periodontal disease.
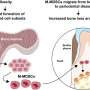
In healthy bones, stromal osteoblast on the outer surface of a bone lay down new bone material, whileosteoclastoriginated from hematopoietic cells break down the old bone for resorption of minerals; the balance between their activities sustains bone mass. A protein called RANKL plays a role in maintaining that balance and, thus, in how bone gets successfully remodeled. The hormone-like PGE2(prostaglandin E2) molecule, naturally produced by osteoblasts, upregulates RANKL during gum inflammation. Alterations in the production of PGE2, and therefore RANKL, would affect bone loss and gain.
Using osteoblasts and bone marrow cells from mice, plus a synthetic molecule analogous to dsRNA, the study authors experimented with exposure of the cells to dsRNA. They observed that the dsRNA clearly induced the differentiation of more osteoclasts, the cells that break down bone. The dsRNA caused osteoblasts to produce more of the hormone-like PGE2that in turn upregulated RANKL and stimulated osteoclasts to differentiate. So, the osteoblasts, through interactions with the dsRNA molecules, sent cellular signals that increased the production of the bone-eroding osteoclasts. The dsRNA also made mature osteoclasts survive longer.
More, longer-surviving osteoclasts lead to more adsorption of bone when gums are inflamed from bacterial disease. The study revealed a previously unknown mechanism by which gum disease causes breakdown of bones. Says Inada, "These data suggest that TLR3 signaling in stromal osteoblast controls PGE2production and induces the subsequent differentiation and survival of mature osteoclasts." The stromal osteoclasts lead to inflammatory resorption of bones anchoring the teeth. Knowing that the inflammation leading to bone damage in periodontitis can be set off by dsRNA introduced via the bacteria or an accumulatedimmune cellsin tissues is a leap forward in combatting the effects of gum disease.
Looking ahead, the researchers plan to further examine how dsRNA—by signaling immune system receptors on stromal osteoblasts to make more PGE2—contributes to progression of periodontitis over time. Understanding the underlying mechanisms is the foundation for novel development of drugs to prevent bone loss from gum disease.