全外显子组测序预测患者是否对癌症免疫治疗有反应
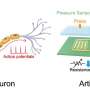
 Combine immunotherapy whole-exome sequencing datasets. 2) Identify mutated genes and pathways. 3) Test candidate genes and pathways for response association. 4) Predictive framework for therapy response (determining Responder or Non-responder). Credit: Sanjana and Imieliński Labs")
免疫疗法,如免疫检查点抑制剂,已经改变了晚期癌症的治疗。与杀死癌细胞的化疗不同,这些药物帮助人体的免疫系统自己发现并摧毁癌细胞。不幸的是,只有一小部分患者对免疫检查点抑制剂有长期反应,而且这些治疗成本高,而且有副作用。
研究人员已经开发了一种两步方法,使用全外显子组测序来瞄准基因以及预测癌症患者是否会对免疫疗法.这项研究发表在自然通讯由纽约大学、威尔康奈尔医学院和纽约基因组中心的研究人员进行的研究,说明了使用全外显子组测序如何比目前的实验室测试更好地预测治疗反应。
“我们能更好地预测谁将受益于免疫疗法吗?科学家们已经开发出各种生物标志物,帮助预测免疫疗法的治疗反应,但对一个强大的、临床实用的预测模型的需求仍然没有得到满足,”纽约大学生物学助理教授、纽约大学格罗斯曼医学院神经科学和生理学助理教授、纽约基因组中心的核心教员、该研究的共同高级作者Neville Sanjana说。
一些生物标志物,包括年龄,肿瘤类型,以及在肿瘤中发现的突变数量癌症细胞被称为肿瘤突变负担,已经知道与免疫治疗的反应相关。通过分析几百个基因计算出的肿瘤突变负担是最可靠的预测因子,通常用于确定患者是否有资格使用免疫检查点抑制剂。
如果科学家们观察我们更大一部分的基因,这是否有助于更好地预测哪些患者会对免疫疗法产生反应?全外显子组测序是一种对编码蛋白质的基因组部分(约2万个基因,占基因组的2%)进行测序的方法,以寻找可能与疾病有关的突变。
虽然全外显子组测序在癌症治疗中没有广泛应用,但最近一些免疫疗法的研究已经开始包括测序。这些研究规模很小,但加在一起可以帮助阐明基因组因素与患者对免疫治疗的反应之间的关系。
研究人员结合了之前六项针对黑色素瘤、肺癌、膀胱癌和头颈癌患者的免疫治疗研究的数据。所有接受免疫检查点抑制剂(抗pd -1或抗ctla -4)治疗的参与者均可获得全外显子组测序。
但即使将这六项研究结合起来,患者的数量——总共319人——仍然相对较少。
“只有几百人的小型研究的问题是患者数量和整个外显子组测序中大量基因测序之间的不匹配。理想情况下,我们的数据集应该有更多的患者,而不是基因,”Sanjana实验室的研究生、该研究的第一作者Zoran Gajic说。
为了解决这个问题,研究人员采用了一种名为鱼钩它将导致癌症的突变与背景突变区分开来,背景突变是指偶然发生但与癌症无关的突变。该模型校正了一系列影响背景突变率的因素,例如,调整基因的大小,因为较大的基因更有可能发生突变。
利用这个模型,研究人员采用了两步方法:首先,他们查看所有患者的测序,以找到任何比他们预期的突变负担更高的基因,并根据基因大小等基因组因素进行调整,或者某个特定的DNA片段是否是一个倾向于积累更多突变的已知热点。这产生了6个可疑的高突变负担基因。
接下来,研究人员确定这六种基因中的任何一种是否在对免疫治疗有反应或没有反应的人身上得到了丰富。其中两种基因——kras(肺癌中经常突变的基因)和BRAF(黑色素瘤中最常见的突变基因)——在对免疫治疗有反应的患者中富集。相比之下,另外两个基因tp53和bclaf1在那些对免疫治疗没有反应的人体内富集。BCLAF1还没有得到很好的研究,但这些发现表明,BCLAF1突变的患者对免疫检查点抑制剂的反应不太可能。
使用相同的两步方法收集称为通路的基因,研究人员确定某些通路(MAPK信号,p53相关和免疫调节)也预测免疫检查点抑制剂反应。
然后,他们将这四种基因和三种途径与年龄、肿瘤类型和肿瘤突变负担等其他预测变量结合起来,创建了一种他们命名为癌症免疫治疗反应分类器(CIRCLE)的工具。CIRCLE能够比单独预测肿瘤突变负担更好地预测约11%的免疫治疗反应。CIRCLE还能够准确预测免疫治疗后癌症的生存期。
“这些结果表明,使用更广泛的诊断方法,如全外显子组甚至全基因组测序,可能会显著提高我们预测谁将对免疫治疗产生反应的能力——本质上,表明更多的数据确实有助于更好地预测治疗反应。”Weill Cornell医学院计算基因组学副教授、病理学和检验医学副教授、纽约基因组中心的核心教员、该研究的共同高级作者Marcin imielizynski说。
为了验证他们的方法,研究人员对另外165个数据进行了CIRCLE测试癌症患者使用全外显子组测序,他们接受了免疫治疗,并发现CIRCLE捕获了仅从肿瘤突变负担中获得的预测信息。
未来的研究将涉及在更大的患者数据队列上测试CIRCLE,因为研究人员预计,该模型将通过来自数千名患者的数据而不是数百名患者的数据来改进。他们还希望通过更大的队列,他们可以开始梳理出哪些患者可能对不同的免疫疗法有反应,因为可用的治疗方法越来越多。
“我们设想这种两步走的方法和使用全外显子组测序将为癌症免疫治疗提供更好的预后工具铺平道路,”Sanjana说。
进一步探索