ALS遗传形式的研究药物改善了疾病的分子体征
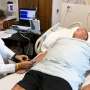
 patient at Washington University School of Medicine in St. Louis. Miller led an international phase 3 clinical trial for a rare, inherited form of ALS. The trial, sponsored by the pharmaceutical company Biogen, showed that an investigational drug, known as tofersen, reduced molecular signs of disease, but at six months did not improve motor control and muscle strength. However, Miller and colleagues found evidence that longer-term use of the drug may help stabilize muscle strength and control. Credit: Huy Mach")
根据结果,一种研究性药物用于治疗一种罕见的遗传性肌萎缩性侧索硬化症(ALS)减少了致命,瘫痪疾病和遏制神经退行性的分子迹象,但是六个月后,该药物并没有改善运动控制和肌肉的强度。从圣路易斯华盛顿大学医学院研究人员领导的第三阶段临床试验。
但是,研究人员发现证据表明,长期使用该药物可能有助于稳定肌肉力量和控制,这是研究人员称为鼓励的发现。该试验是由实验药物制造商Biogen制药公司Biogen赞助的。数据于9月22日发布新英格兰医学杂志。
试验的参与者携带一个名为的基因中的突变SOD1这创建了以同名蛋白质的错误折叠版本,导致ALS,也称为Lou Gehrig氏病。
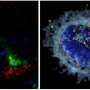
该试验表明,该药物(称为Tofersen)降低了SOD1的水平以及神经丝蛋白,这是神经系统损害的分子标记。在安慰剂控制的一部分结束时,为参与者提供了接收Tofersen的选择,作为开放标签扩展名的一部分,该扩展名将持续长达4。5年。开放标签扩展的建立创造了两组参与者:从一开始就一直在Tofersen上的人,以及那些在开始Tofersen之前接受了安慰剂六个月的人。延长六个月的临时分析显示,早期和晚期开始者的运动功能存在显着差异。一年服药后,一些参与者表现出稳定肌肉力量研究人员说,控制这是一个以不断下降的特征的疾病,这是一个了不起的发现。
开放标签的扩展正在进行中,研究人员继续监视参与者的运动功能。7月,食品药品监督管理局接受了Biogen对Tofersen的新药物申请,作为与与之相关的ALS的治疗SOD1突变。
首席研究员蒂莫西·M·米勒(Timothy M. Miller)博士说:“这是为与SOD1相关的ALS寻找治疗疗法的激动人心而充满希望的一步。医学ALS中心。“我们看到明确的证据表明该药物减慢了起始因素的减慢 - ASOD1突变 - 以及神经退行性疾病过程。我们在六个月时没有看到大量的临床改善,但是在较长时间点的功能和强度稳定表明,人们可能需要花费时间来治愈已经造成的损害。绝大多数拥有ALS的人都经历了不懈的渐进下坡课程,因此在开放标签扩展期间的功能稳定确实是显着的。”
美国约有20,000人与ALS生活在一起。致命的疾病杀死了控制步行,进食和呼吸的神经细胞。诊断后五年以上,很少有人生存。
大约2%的ALS病例是由突变引起的SOD1。Tofersen是一种反义寡核苷酸,这是一种基于DNA的分子,它会干扰建立蛋白质的遗传指令。该分子旨在阻断SOD1蛋白的产生。
第三阶段试验是在10个国家 /地区的32个地点进行的,其中包括108名ALS患者SOD1突变。在24周的时间内,将三分之二(72)的参与者随机分配给八剂Tofersen,直接在其脊髓周围的流体中施用。其余36人接受了八剂安慰剂。所有参与者在入学时进行评估,并在28周内进行衡量运动功能在四个领域:吞咽和讲话;呼吸;精细的运动技能;和总体运动技能。他们还提供了脊髓液样品,因此研究人员可以测量与ALS相关蛋白的水平。
当试验结束时,有95名参与者继续进行开放标签扩展。扩展名中的所有参与者都会收到Tofersen。扩展参与者和研究人员都不知道在试验期间谁接受了Tofersen或安慰剂。
“在NEJM让ALS社区的兴奋和希望可以放慢或阻止疾病进展的疗法。”药物有可能通过长期使用来稳定肌肉功能来改善患有SOD1-AL的人的生活质量,这是一个极其有希望的发展。”
华盛顿大学神经病学教授罗伯特·布塞利(Robert Bucelli)是该大学ALS中心的联合主任。作为华盛顿大学临床试验的现场负责人,他照顾10名参与者。
Bucelli说:“我们网站的大多数正在进行的参与者都恢复了和/或维持了许多日常生活活动,我们的考试和力量测量证实了他们的改善,稳定或两者的历史。”“作为一名神经肌肉临床医生,亲眼目睹这一亲戚的特权改变了我对这种和其他相关的,毁灭性神经退行性疾病的看法。”
尽管该试验的结果仅适用于因突变引起的ALS的人SOD1,他们可以告知研究,可能使其他形式的疾病受益。
米勒说:“我一直认为ALS是一种可治疗的疾病。”“这是我整个职业的基础,即神经退行性疾病(包括ALS)的假设无需致命。如果您查看这项研究的较晚时间,它们显示出大量的神经变性,患有患者的神经变性很大SOD1-als。我认为这对于具有任何形式的ALS的人来说是希望的消息。它告诉我,如果我们找到正确的疗法,我们可以改变疾病的进程。我们只需要找到正确的疗法即可。”