删除绑定网站一个致癌基因可以减缓癌细胞的生长
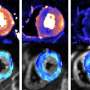
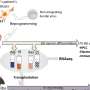
 and desired mutation (orange). In addition, the HDR templates harbor sequence tags that can be identified by Illumina sequencing of the targeted locus, enabling lineage tracing of the edited clones and creating a large number of internal replicates in each experiment. The sequence tags are generated by mutating nucleotides flanking the region of interest with the probability of 24%, a strategy that typically introduces 2–3 mutated nucleotides (indicated with red diamonds; Extended Data Fig. 1), leaving most of the flanking sequence intact, as demonstrated by the position weight matrices. b, Experimental strategy using a mixture of HDR template libraries harboring the original and mutated sequences for the same target. The abundance of each HDR template in the cell population is analyzed from the sequence tags after different assays and compared to respective baseline: cellular fitness (gDNA at day 8/day 2), TF binding (chromatin-immunoprecipitated DNA/input DNA) and mRNA expression (mRNA abundance/respective gDNA). c, The number of possible sequence variations with zero (n = 1), one (n = 30), two (n = 405) and three (n = 3,240) flanking mutations when the sequence tags are created by mutating ten nucleotides with the probability of 24% and their abundance in the HDR template library analyzed from read counts in ChIP input sample of the edited SHMT2 E-box locus. The box plots indicate the median read count with upper and lower quartiles, and the whiskers extend to 1.5 times the interquartile range. The number of sequence tags recovered in each experiment is shown in Supplementary Table 3. d, The effect of E-box mutation at the RPL23 gene promoter on fitness of HAP1 cells shown by read count ratios for mutated/original sequences for each cell lineage pair harboring identical sequence tags with one flanking mutation. Credit: Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01444-6")
研究人员关注MYC癌基因的影响揭示了新信息的因素调节肿瘤细胞的生长。MYC促进细胞增长的重要基因的表达,它是活跃在超过半数的人类癌症。然而,目前还没有药物可以抑制MYC函数,因为它的蛋白质结构并不适合治疗的目标。选择预防MYC活动会抑制MYC目标基因的功能。在最近的研究中,研究人员芬兰赫尔辛基大学属于学院的卓越中心的肿瘤遗传学的研究已经确定了目标的MYC基因负责经济增长的促进作用。
“在一个健康的组织,细胞生长和扩散是严格控制的过程。在癌症的发展,细胞逃避这些控制机制和肆意地生长,”高级研究员解释说Paivi Pihlajamaa。
研究人员发现MYC癌基因的基因组结合位点的监管区域目标基因和显示,删除这些结合位点DNA减慢细胞生长。这项研究最近发表在自然生物技术。
“我们的研究结果表明,细胞DNA变化很小,如修改单个基因调控元素,可以显著影响细胞的增殖率,”证实了Pihlajamaa。
这些发现可能在未来许多癌症患者受益。
“更好的理解机制,控制细胞的增长可以帮助确定目标可以被新癌症药物,”Pihlajamaa告诉。
进一步研究新方法的好处
本研究的另一个主要影响来自由研究人员开发的一种新方法。重要的是,它使测量的影响变化的小DNA元素在细胞增长强劲和精确。
“未来的研究方法是非常有益的,因为它可以用于研究各种变异如何影响其他细胞增殖和属性,“组长Jussi Taipale教授解释道。
该方法是基于所谓的遗传剪刀,即CRISPR-Cas9系统,最近已成为生物医学研究的一个重要工具。ob欧宝直播nba系统吸引了很多关注,因为它使基因组编辑效率和精度高。
基因组编辑过程,然而,诱发DNA损伤反应,影响细胞的生长。因此,研究人员必须能够区分的影响感兴趣的突变引起的基因组编辑过程本身。
“我们的方法解决了这个问题通过使用一个实验性的策略的细胞发生了基因组编辑直接与对方相比,和未经审查的野生型细胞被丢弃的分析,“Taipale告诉。
“这是一个功能强大的方法,可以在阐明产生重大影响的过程控制细胞增长在未来,“Taipale总结道。
更多信息:Paivi Pihlajamaa et al,竞争精密CRISPR方法识别转录因子结合位点的健身效果,自然生物技术(2022)。DOI: 10.1038 / s41587 - 022 - 01444 - 6